-
A Synthetic and Mechanistic Investigation of the Chromium Tricarbonyl-Mediated Masamune–Bergman Cyclization. Direct Observation of a Ground-State Triplet p-Benzyne Biradical

K.E.O. Ylijoki, S. Lavy, A. Fretzen, E.P. Kündig, T. Berclaz, G. Bernardinelli and C. Besnard
Organometallics, 31 (15) (2012), p5396-5404


DOI:10.1021/om300427j | unige:22151 | Abstract | Article HTML | Article PDF
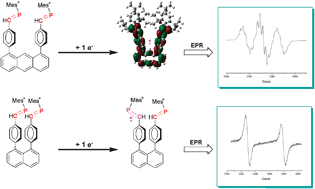
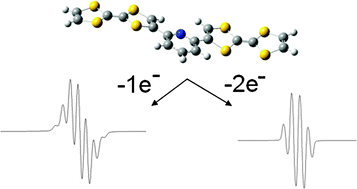
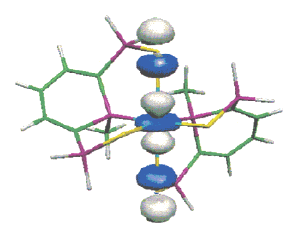
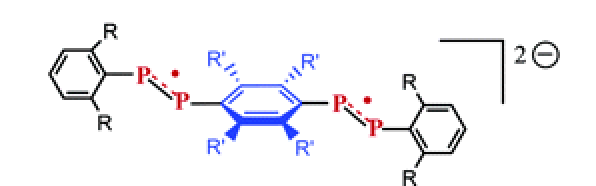
A new room-temperature chromium tricarbonyl-mediated cycloaromatization of enediynes is reported. The reaction occurs with both cyclic and acyclic enediynes in the presence of [Cr(CO)3(η6-naphthalene)] and both a coordinating solvent and a hydrogen atom source, providing chromiumâarene complexes in reasonable yield and good diastereocontrol. The mechanism of the reaction has been probed through DFT computational and spectroscopic methods. These studies suggest that direct C1âC6 bond formation from an η6-enediyne complex is the lowest-energy path, forming a metal-bound p-benzyne biradical. NMR spectroscopy suggests that enediyne alkene coordination occurs in preference to alkyne coordination, forming a THF-stabilized olefin intermediate; subsequent alkyne coordination leads to cyclization. While biradical quenching occurs rapidly and primarily via the singlet biradical, the triplet state biradical is detectable by EPR spectroscopy, suggesting intersystem crossing to a triplet ground state.